Ученые разработали метод, который позволяет находить стабильные геометрии соединений, упускаемые при молекулярном моделировании. При тестировании на 60 потенциально биологически активных соединениях для почти половины молекул алгоритм обнаружил до 28 конформаций (геометрий), не выявленных существующими методами. Новый подход, сочетающий в себе квантово-химические расчеты и машинное обучение, анализирует геометрические варианты молекул, построенные другими методами конформационного поиска, и находит недостающие всего за 20–30 попыток. Инструмент может использоваться для поиска и разработки фармацевтических соединений и новых катализаторов. Результаты исследования, поддержанного грантом РНФ, опубликованы в Journal of Chemical Information and Modeling.
Большинство молекул могут принимать несколько пространственных форм — геометрий, или конформаций, — из-за вращения частей молекулы относительно друг друга. Каждая конформация имеет свои химические и физические свойства, поэтому для предсказания свойств соединения с помощью квантово-химического моделирования необходимо учитывать все его возможные геометрии. Важно отметить, что всего одна пропущенная конформация может качественно исказить результаты моделирования, сделав их бесполезными (а в некоторых случаях вредными) для создания целевого вещества. Однако даже самые точные современные методы могут упускать наиболее устойчивые конформации молекул.
Ученые из Института органической химии имени Н. Д. Зелинского РАН (Москва) и Московского государственного университета имени М. В. Ломоносова (Москва) создали метод на основе искусственного интеллекта, который помогает находить пропущенные геометрии в наборах конформаций молекулы.
Новый алгоритм основан на методе машинного обучения — гауссовских процессах. На первом этапе модель запоминает данные об энергии вращения частей молекулы друг относительно друга, которые ученые получают с помощью квантово-химических расчетов. Затем, в отличие от традиционных подходов, программа не только фокусируется на поиске самой выгодной конформации (глобального минимума), но обращает внимание и на недоисследованные области конформационного пространства. Это позволяет модели определять, какие особенности структуры могут быть выгодны, даже если они не обнаружены с помощью ранее проведенного конформационного поиска. Еще одно преимущество подхода состоит в том, что он требует всего 20–30 попыток для нахождения пропущенных молекулярным моделированием конформаций, что делает его применимым для крупных молекул с большим количеством заместителей (фрагментов из одного или нескольких атомов).
«Обычно в науке применяются классические методы машинного обучения, разработанные для использования в ситуациях, когда данных для обучения много. Часто же задачи, возникающие в химии, не могут удовлетворить этим условиям, поскольку получение данных в них сопряжено с затратными экспериментами. В связи с этим кажется перспективным применение байесовских методов — подходов, при которых модель самостоятельно оценивает свою уверенность в предсказаниях. Это преимущество позволяет успешно применять такие методы даже в ситуациях, когда данных очень мало — всего десятки измерений. Именно при помощи байесовских методов, к которым относятся гауссовы процессы, нам и удалось достигнуть полученного результата», — рассказывает участник проекта, поддержанного грантом РНФ, Иван Беспалов, студент химического факультета МГУ, сотрудник группы теоретической химии Института органической химии.
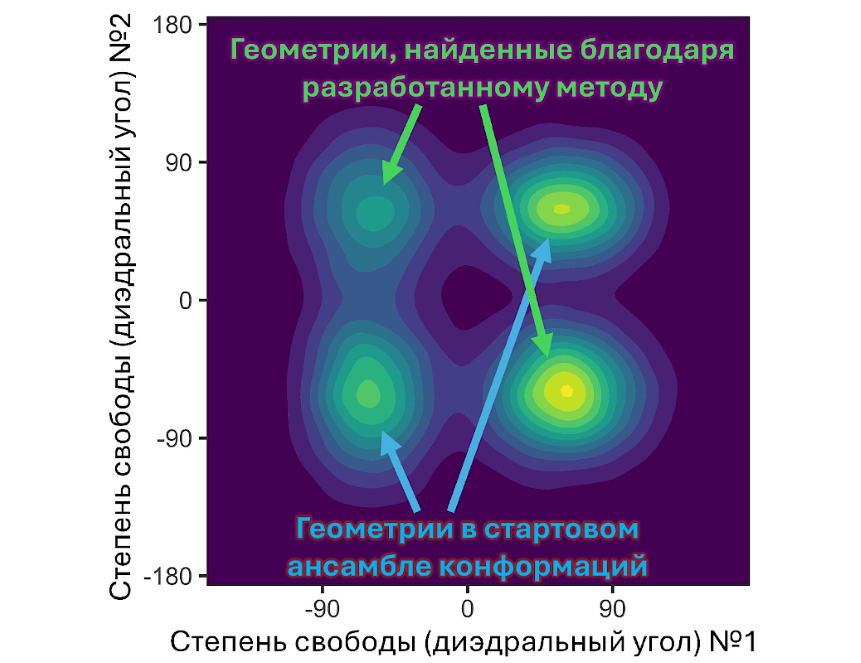
Авторы протестировали алгоритм на 60 биологически значимых молекулах, таких как пептиды и лекарственные соединения, используя данные о конформациях, полученные с помощью одного из наиболее надежных и современных методов конформационного поиска — CREST. Оказалось, что новый подход обнаружил конформации, которые упустил CREST, для 24 из 60 молекул. Причем в отдельных случаях метод нашел до 28 новых конформаций. При анализе соединений, содержащих амидные фрагменты — азотсодержащие химические группы, встречающиеся во всех белках и пептидах, — алгоритм во всех случаях нашел пропущенные CREST энергетически выгодные формы.
«Разработанный нами метод позволяет существенно повысить надежность молекулярного моделирования и увеличить скорость поиска новых стабильных органических и металлоорганических веществ с заданными свойствами, которые потенциально могут стать, например, лекарственными препаратами или новыми катализаторами. Он станет важным шагом к автоматическому молекулярному моделированию, которое позволит надежно получать достоверные результаты с минимальным участием человека. Сейчас мы продолжаем работу над другими цифровыми инструментами, комбинирующими физику и искусственный интеллект, которые должны закрыть другие проблемы, отделяющие нас от этой цели», — подводит итог руководитель проекта, поддержанного грантом РНФ, Михаил Медведев, кандидат физико-математических наук, старший научный сотрудник группы теоретической химии Института органической химии.
Подписывайтесь на InScience.News в социальных сетях: ВКонтакте, Telegram, Одноклассники.





